|
|

|

|
|

|
뉴스
의약품 품질시스템 도입 등 GMP 종합 개편안 발간
식약처, 2월~10월 시행한 주요 제도 개선사항 현장 적용 위해 마련
박선혜 기자 │ loveloveslee@yakup.com

입력 2020-12-02 09:41 수정 2020.12.02 09:50




식품의약품안전처(처장 김강립)는 올해 추진한 의약품 제조 및 품질관리기준(GMP)에 관한 평가 방법 개편사항 등을 종합한 안내서를 발간했다.
이번 안내서는 올해 2월부터 10월까지 시행한 주요 제도 개선사항을 제약 현장에서 원활하게 적용할 수 있도록 마련됐으며, 업계의 이해도를 높이기 위해 그간의 주요 질의·답변 내용을 추가했다.
주요 내용으로 동일한 제조공정으로 위탁생산하는 전문의약품에 대한 자료제출 요건 강화를 위해 전공정 위탁제조 품목(전문의약품) GMP 평가 자료제출 의무화했다.
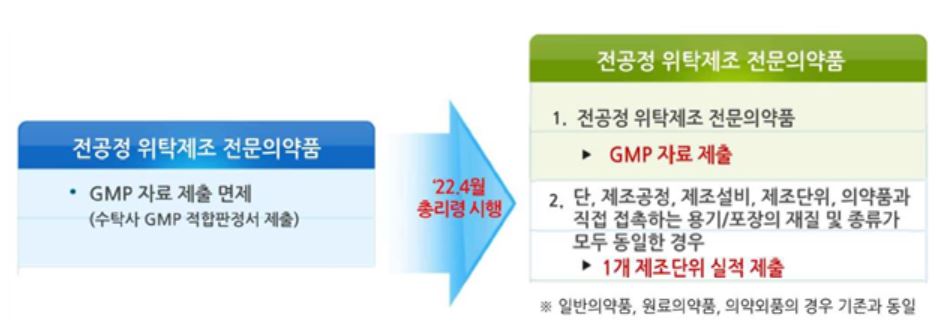
|
또한 전주기 관리를 위한 ‘의약품 품질시스템(Pharmaceutical Quality System)’을 도입
해 의약품 개발부터 생산 및 판매종료까지 지속적 개선을 촉진하는 관리 시스템으로 의약품 제조 및 품질관리기준과 품질위험관리를 포함한다.
이를 통해 품질 결함에 대한 조사를 위해 유통 중인 제품을 수거하는 경우에는 이를 회수로 간주하지 않도록 개선하고 위탁자 및 수탁자 간 책임과 기준 등에 대한 의약품 제조 및 품질관리 기준의 명확화할 수 있다.
더불어 식약처는 의약품 허가신청 자료 및 품질관리의 신뢰성 향상을 위한 ‘의약품 제조·수입업체 데이터 완전성 평가지침’을 마련했다.
‘의약품 제조업체 데이터 완전성 평가지침’에 적합하도록 당해 업체의 품질관리기준서 등 4대 기준서를 반영한다. 적용대상은 2021년 1월 1일 이후 식약처에 허가 신청하는 신약(신물질 원료의약품 포함)에 한정(행정 지시일 이전 작성된 자료는 적용대상에서 제외)한다.

|
‘의약품 제조업체 데이터 완전성 평가지침’에 적합하도록 당해 업체의 품질관리기준서 등 4대 기준서를 반영한다. 적용대상은 2021년 1월 1일 이후 식약처에 허가 신청하는 신약(신물질 원료의약품 포함)에 한정(행정 지시일 이전 작성된 자료는 적용대상에서 제외)한다.
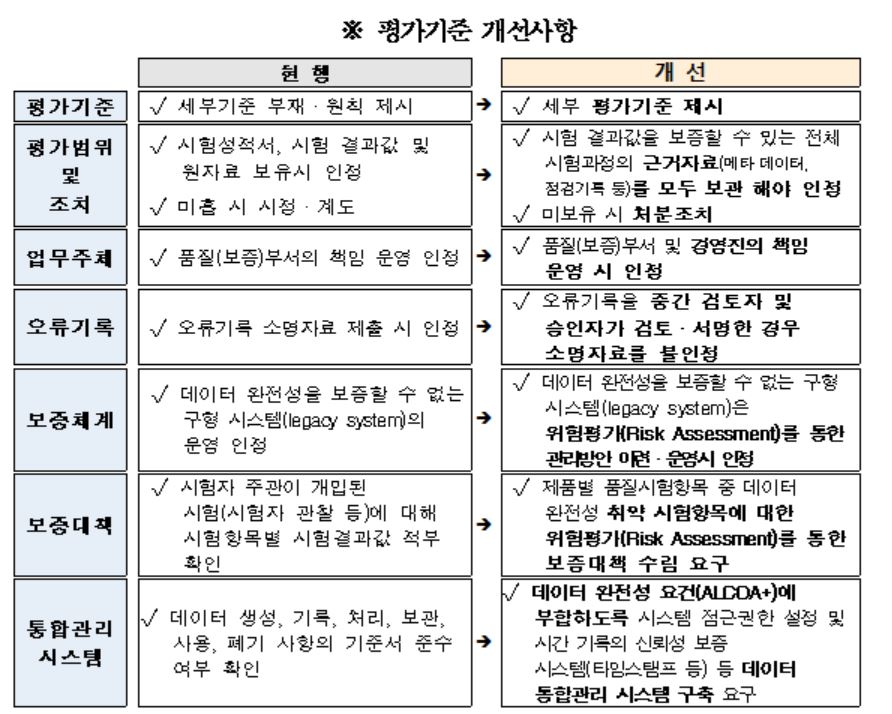
|
더불어 코로나19 확산 및 장기화에 따라 단계적으로 시행하는 수입의약품 GMP 평가 도 개편했다. ‘코로나 19’ 상황 종료시까지 접수되는 허가 또는 등록 신청 건(기 접수 품목 및 변경 포함)에 대해 평가방안(1~3차 종합)을 적용할 계획이다.
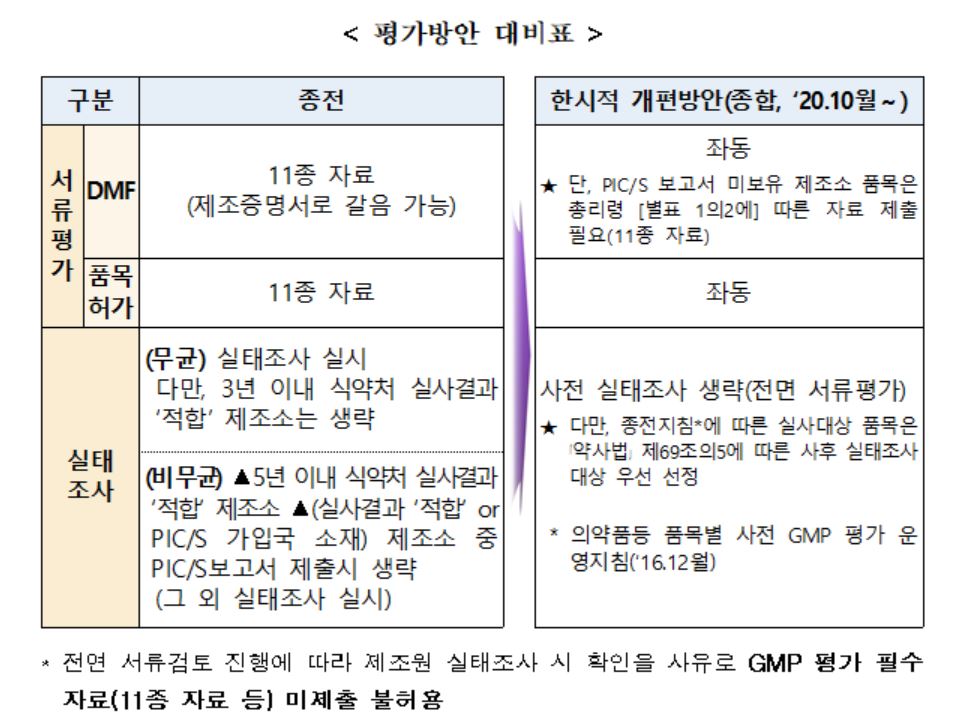
|
식약처는 앞으로도 국내 제약업계에서 국제적 수준의 제조 및 품질관리기준(GMP)을 도입·활용하도록 하여 보다 우수한 품질의 의약품을 국민께 공급하기 위해 노력하겠다고 밝혔다.





전체댓글 0개
등록된 댓글이 없습니다.

오늘의 헤드라인
-
01 팍스로비드 반품 허용한 화이자...약사회 요... -
02 제넨바이오, 회생절차 폐지 신청 -
03 에이프로젠바이오로직스, 1Q '적자 늪'서 여... -
04 에이비엘바이오, 바이오 USA 출격…글로벌 파... -
05 진매트릭스, 중기부 ‘글로벌 강소기업 1000+... -
06 포리바이오, 정제 공정 고도화... 원료 생산... -
07 상장 제약바이오 1분기 평균 매출, 코스피 1... -
08 리가켐바이오,노바락바이오테라퓨틱스와 항... -
09 샤페론, 염증복합체 억제 아토피 치료제 FDA... -
10 인피니트헬스케어 상근 감사," 행동주의 주...