|
|

|

|
|

|
뉴스
의약품 제조과정 전자기록·전자서명 신뢰성 확보 방안은?
[기고](주)피티케이 이시훈 차장, 미국 FDA ‘21 CFR Part 11’ Checklist
김정일 기자 │ jikim@yakup.com

입력 2017-06-01 11:27 수정 2017.06.02 16:41





|
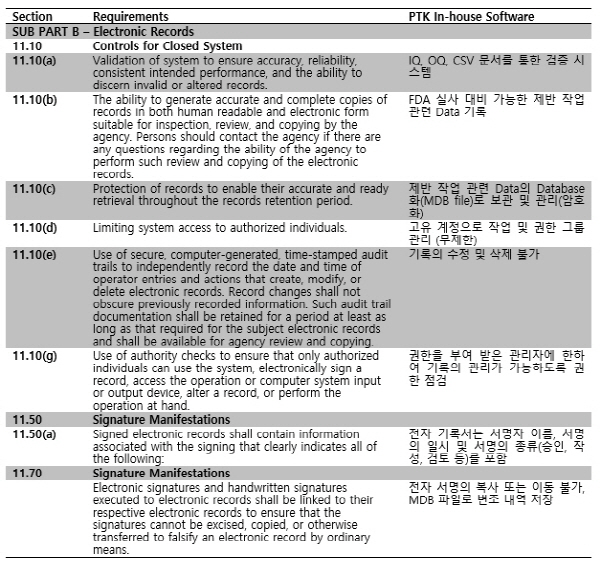
그 중 열한 번째 항목인 Part 11은 전자기록과 전자서명이 기존 기록물(Documents)과 동일한 신뢰성을 확보할 것을 법률적으로 강제하고 있는 조항이다.
신약 개발부터 제조 관리에 이르기까지의 모든 공정을 기록 관리해야 하는 제약 산업 분야의 특성상, 업계에서는 전체 공정을 포괄하는 방대한 양의 종이 기록물이 생성 및 관리되어왔다.
이후 관련 장비에 컴퓨터를 적용하는 게 일반화되면서, 기존의 종이 기록물을 전자적 파일로 효과적으로 대체할 수 있는 기술이 요구되었고, 자연적으로 이러한 기술들을 관리할 법안이 체계화되었으며, 이것이 Part 11의 탄생 배경이 된다.
법규 발표 이후, 제약회사들은 법규를 준수해야 할 ‘당사자’가 되었지만 적절한 대응책을 준비하지 못한 채 혼란스러워 했을 뿐 아니라 추가로 드는 비용 부담에 대한 불만을 토로하기도 했다.
이 가운데 2003년 ‘FDA Guidance for Industry Part 11, Electronic Records; Electronic Signatures — Scope and Application’가 발표된다.
이것은 Part 11에 대해 FDA가 어떤 기준을 가지고 법규를 실행할 것인지 관련 업체들에게 좀 더 명확히 설명하는 가이드라인으로써 그 동안 모호했던 법규를 구체적으로 설명한 중요한 문서이며 업계에서는 이를 기준으로 Part 11에 대응하는 시스템을 구축하고자 노력하고 있다.
2007년 FDA는 병원 등의 의료시설에서 사용하는 컴퓨터 시스템에 대한 가이드라인을 발표하기에 이르렀고 관련 업계는 필요한 시스템을 갖춰 가이드라인 요구에 맞게 전자기록과 서명 Audit trail에 대한 FDA 현장 조사에 대응하기 시작했다.우리나라 식약처도 2010년 임상시험 자료관리에 대한 전자기록 가이드라인을 발표하는 등 미국 FDA 정책에 발맞추어 관련 법규에 대한 관리를 강화해 나가고 있는 실정이다.
현재 해당 Part 11에서 명시하는 요구사항에 대한 업계의 이해도는 꽤 높은 편이고, 이를 바탕으로 약품 제조 관련 가이드라인 발표 역시 어느 정도 임박한 상태라 할 수 있다.
많은 제약사들이 신규 장비 구매시 컴퓨터 시스템 장착을 적극적으로 검토하고 있어 Part 11 요구 사항에 대한 업계 전반의 대응력이 높아지고 있는 실정이다. 그러나 다른 한편으로, 준비 비용이 여전히 부담스러우며, 시스템이 Part 11에 얼마나 부합하는지에 대한 신뢰도에 많은 의문이 제기되는 등 Regulation 대응이 그리 쉽지만은 않은 것도 사실이다.
Part 11은 FDA의 관리 범위에 있는 국가의 의약품 제조업체는 물론 해당 국가에 판매 승인을 얻고자 하는 전 세계의 제약사를 그 대상으로 한다.
미국의 FDA와 같은 역할을 하는 유럽연합의 기구인 EMA(European Medicines Agency)도 Part 11과 동일한 가이드라인인 Annex 11( European Medicine Agency's Guidelines to Good Manufacturing Practice - Annex 11)을 발표했고 이를 통해 EU 국가 내의 제약사에 전자기록과 전자서명의 준수를 요구하고 있다.
시간이 지날수록 제약 산업은 국경이 무의미한 글로벌 형태의 비즈니스로 변화하고 있으며 그러한 변화 속에 제조와 마케팅의 영역은 그 경계가 모호해지고 있다. 법규도 마찬가지다. FDA가 미국 업계에 요구하는 것은 곧 우리도 지켜야 할 의무가 되고 새로운 가이드라인에 대응하기 위한 준비 시간의 사이클은 점차 짧아지고 있다.
일례로 신규 설비 검토 시 작성하는 URS(User Requirement Specification)의 첫 부분에서 21 CFR 210/211 이라든지 EudraLex 의 조항들은 이제 필수 항목이 되었다.
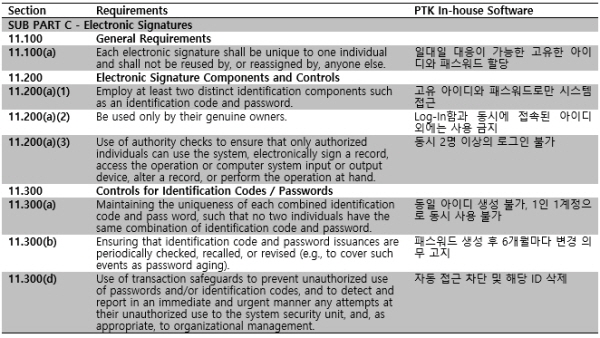
Part 11을 준수한다는 것은 FDA 혹은 식약처가 기존 GMP(Predicate Rule)에서 규정했던 모든 형태의 서류들을 전자 포맷으로 제출해야 한다는 뜻이다. 종이서류를 전자적 방법으로 보내는 것은 허용이 되지 않는다.
예를 들어 Part 11을 준수하는 제약회사 생산현장에서 타정 작업을 시작한다고 하자. 담당 작업자가 ID/PW를 입력해 타정기에 접속 후, 작업을 시작하면 동시에 실시간으로 타정 조건들(터렛 속도, 피더 속도, 타정압, 경도, 중량, 생산량 등)이 HMI 시스템에 의해 기록된다.
공정관리실에서는 해당 데이터를 사내 MES 네트워크를 통해 관리하고 분석하며 레포트 형태로 출력을 하기도 한다.(ERES : Electronic Record & Electronic Signature)
가끔 데이터 오기에 따른 수정을 해야 할 때도 있는데 그러한 경우에도 원본 데이터는 변경되지 않고 수정한 데이터에 대해서는 누가, 언제, 무엇을 수정했는지 기록이 남게 된다.(Audit Trail)
전자서명 기능을 통해 각 권한 별로 승인되고 축적된 기록들은 모두 통합 데이터베이스에 저장이 되어 추후 식약청의 요구가 있으면 전자적 방법으로 제출된다.
각 회사마다 고유의 생산관리 시스템을 가지고 있고 관리하는 방식도 다르기 때문에 설비를 구매할 때 컴퓨터 시스템의 안정성, 호환성, 확장성이 중요한 선택기준이 된다.
아래의 체크리스트에 Part 11의 핵심 세부 규정 별 장비 시스템의 대응을 요약해 놓았다.
<참고문서>
1. FDA Title 21 CFR Part 11:Electronic Records; Electronic Signatures; Final Rule (1997)
2. FDA Guidance for Industry Part 11, Electronic Records: Electronic Signatures – Scope and Application (2003)
3. Guidance for Industry Computerized Systems Used in Clinical Investigations (2007)
|
|





전체댓글 0개
등록된 댓글이 없습니다.

오늘의 헤드라인
-
01 한국유니온제약 상폐 수순…정리매매 돌입 속... -
02 "공정 불순물 관리 핵심 ‘HCP’ 분석·제어 전... -
03 에피바이오텍, 동종 모유두세포 치료제 핵심... -
04 트럼프,의약품 관세 부과...한국산 의약품 1... -
05 큐라클, CU01 당뇨병성 신증 임상2b상 효능·... -
06 LG화학, 모치다제약 자궁내막증 치료제 ‘디... -
07 [2026 기대 신약 TOP 10] ② 비만 치료제 '올... -
08 [영상] KOREA PACK & ICPI WEEK 2026, 제조 ... -
09 깐깐해지는 의약품 제조 규제… 제약 제조 혁... -
10 [최기자의 약업위키] 자궁내막암 면역항암제...