|
|

|

|
|

|
뉴스
미국 FDA, 임상시험계획 신청·바이오시밀러 승인 간소화 추진
일부 1상 임상시험,위험 기반 신속 IND 경로 새로 만들고
바이오시밀러 승인시 1개 부서서 검토..승인 효율성 높여
바이오시밀러 승인시 1개 부서서 검토..승인 효율성 높여
이권구 기자 │ kwon9@yakup.com

입력 2026-04-07 15:42 수정 2026.04.07 15:52




|
미국 FDA가 임상시험계획 승인 신청 간소화 및 바이오시밀러 승인 간소화 등을 추진한다.
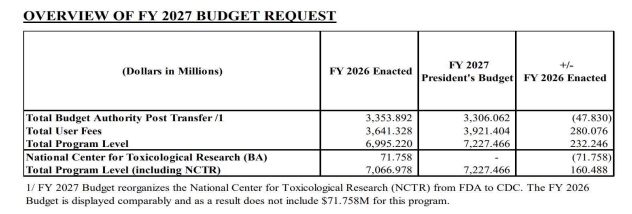
한국바이오협회 바이오경제연구센터 7일 ‘이슈 브리핑’에 따르면 3일 FDA는 2026년 10월 1일부터 시작되는 2027년 회계연도 예산요구서를 통해 2027년 예산으로 전년보다 1억 6천만 달러가 증가한 72억 2천 7백만달러를 요청했다. 이 예산은 정부예산 33억 6백만 달러, 기업들이 내는 사용자수수료(39억2천1백만 달러)로 구성된다.
FDA는 예산을 통해 임상시험계획 승인신청 간소화 및 바이오시밀러 승인 간소화 등을 추진할 계획이라고 밝혔다.
FDA에 따르면 일부 1상 임상시험에 대해, 기존 전임상 데이터가 검증된 NAMS(새로운 대체시험법, new approach methodologies) 방법으로 규제 기준을 잠재적으로 충족할 수 있는 경우, 선택적 위험 기반 신속 IND 경로를 새로 만들 계획이다. 기존의 전통적 IND 경로를 대체할 수 있는 이 새로운 경로는 안전성과 윤리적 기준을 유지하면서 중복적이고 시간 소모적인 요구사항을 줄이는 것을 목표로 한다.
센터는 현재 중국과 호주에서 초기 전임상 및 1상 활동이 증가하고 있다는 점에서, 이러한 정책은 현행 패러다임에서 진입 장벽이 더 높은 소규모 바이오기업에 특히 중요하다고 분석했다. 실제 미국은 전임상 작업과 임상시험 승인 과정 때문에 더 긴 일정과 더 큰 규제 부담을 갖고 있다.
센터는 “이 정책은 미국 기반 1상 임상 프로그램을 시작하는 가속화된 경로를 만들고, 인간 대상 1상 연구를 지원할 수 있는 충분한 전임상 데이터를 갖춘 약물의 규제 부담을 줄여 시장 경쟁을 촉진하며, 약물 개발 비용을 낮춰 대통령의 제약산업 리쇼어링 및 온쇼어링 목표를 지원할 것“이라고 진단했다.
또 FDA는 인터체인저블 바이오시밀러 제도를 폐지하고, 비교임상시험 요건을 삭제하며, 바이오시밀러 승인시 2개 부서가 아닌 1개 단일 부서에서 검토토록 해 바이오시밀러 검토 및 승인 효율성을 높일 계획이다.
이 제안은 공중보건법(PHS Act) 제351조를 개정해 상호교체 가능성(interchangeability)에 대한 별도 법정 기준을 더 이상 포함하지 않고, 승인된 모든 바이오시밀러를 각 해당 참조제품(오리지널의약품)과 상호 교체 가능하다고 간주함으로써 바이오시밀러 검토 및 승인 과정을 간소화하는 것이 골자다.
센터는 “이 제안은 미국의 바이오시밀러 프로그램을 현재의 과학적 이해와 더 일치하게 만들고, 승인 시 바이오시밀러와 해당 참조제품의 상호 교체를 허용하는 유럽연합(EU)과 같은 다른 주요 규제 기관들 접근 방식과도 일치하게 해 미국 바이오시밀러 시장을 유럽 시장과 더 경쟁력 있게 만들며 소비자 비용을 낮추는 효과를 가져올 것“이라고 진단했다.





전체댓글 0개
등록된 댓글이 없습니다.

오늘의 헤드라인
-
01 [약업분석] 알테오젠, 매출 2배·영업익 320%... -
02 미국 FDA, 임상시험계획 신청·바이오시밀러 ... -
03 휴로틱스 'H-Medi', 고령자 임상연구 통해 ... -
04 다케다, 전두측두엽 치매 치료제 개발 중단 ... -
05 중동전쟁 불똥 튄 의료현장…정부, '나프타 ... -
06 GC녹십자웰빙, hADM 무세포동종진피 ‘지셀르... -
07 SK바이오팜, AAN 2026서 ‘세노바메이트’ 임... -
08 삼진제약 "월드클래스 프로젝트 선정… 차세... -
09 거점도매 '전면 철회' 못박은 유통…"타협 없다" -
10 아이엠바이오로직스, AACR 2026서 HLA-G 타...